糖原累积病(glycogen1storage1disease,lGSD)是一大类遗传性糖代谢障碍疾病,l主要病因为先天性糖代谢酶缺陷所造成的糖原代谢障碍。美欧的发病率约为1/25000~1/20000。由于酶缺陷的种类不同,l临床表现多种多样。根据临床表现和生化特征,l共分为十三型,l其中以Ⅰ型1GSD最为多见。
【发病机制】
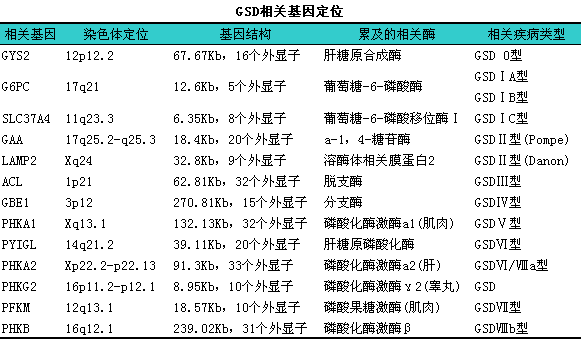
人体糖原合成主要通过四个环节:i①葡萄糖磷酸化;②尿苷二磷酸葡萄糖生成;③α-1,l4糖苷键;④α-1,l6糖苷键。糖原分解是糖原在磷酸化酶作用下,l将α-1,l4糖苷键分解生成葡萄糖1-磷酸,l再由葡萄糖转移酶和分支酶作用,l将α-1,l6糖苷键水解生成游离的葡萄糖。GSD是由于患者缺乏糖原代谢有关的酶,l使糖原合成或分解发生障碍,l导致糖原沉积于组织中而致病。由于酶缺陷的种类不同,l造成多种类型的糖原代谢病,l常见类型见下表,l以葡萄糖-6-磷酸酶(G-6-Pase)缺乏最常见,l其分子病理生理基础是G-6-Pase相关基因突变,l导致G-6-Pase缺乏而引起Ⅰ型GSD。目前已可检测出多种G-6-Pase基因突变,l其中最多见于R83C和Q347X,l约占Ⅰ型GSD的60%。但有地区差异,l中国人群以nt327G→A(R83H)检出频率最高,l其次为nt326G→A(R83C),l因此G-6-Pase基因第83密码子上的CpG似乎是突变的热点。
【临床表现】
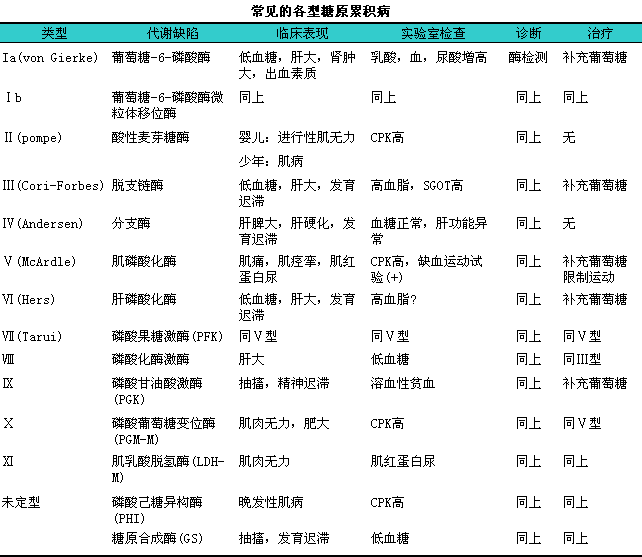
根据酶缺陷不同和糖原在体内沉积部位的不同分为13型,l临床以Ⅰ型糖原累积病最多见,l且Ⅰ、iⅢ、iⅥ、iⅨ型以肝脏病变为主,lⅡ、iⅤ、iⅦ型以肌肉组织受损为主。常见类别及其主要临床表现见下表。
【辅助检查】
1.生化检查11Ⅰ型患者空腹血糖降低至2.24~2.36mmol/L,l血乳酸及糖原含量增高,l血脂酸、i尿酸值升高。
2.白细胞酶的测定11对Ⅲ、iⅣ、iⅥ、iⅨ型病人可能有帮助。
3.糖代谢功能试验
(1)肾上腺素耐量试验:i注射肾上腺素60分钟后0、iⅠ、iⅢ、iⅫ型患者血糖均不升高。
(2)胰高血糖素试验:i0、iⅠ、iⅢ、iⅣ型患者示血糖反应低平,l餐后1~2小时重复此试验,l0、iⅢ型血糖可转为正常。
(3)果糖或半乳糖变为葡萄糖试验:iⅠ型患者在负荷果糖或半乳糖时不能使葡萄糖升高,l但乳酸明显上升。
(4)糖耐量试验:i呈现典型糖尿病特征。
4.肌肉组织或肝组织活检11活检组织作糖原定量和酶活性测定,l可作为确诊的依据,l但损伤性大。
5.分子生物学检测11应用PCR结合DNA序列分析或ASO杂交方法能正确地鉴定88%Ⅰ型糖原累积症患者携带的突变等位基因。基因检测可避免侵害性的组织活检,l亦可用于携带者的检出和产前诊断。
【诊断】
根据典型的病史、i临床体征以及相关的实验室检查结果即可获得临床诊断。而肾上腺素试验或葡萄糖代谢功能试验、i半乳糖代谢功能试验有助于确诊及区分GSD的不同类型。
【治疗】
本病治疗首先应维持患者正常的血糖水平,l以阻断这种异常的生化过程,l从而减轻临床症状。采用适时全静脉营养(TPN)疗法可以纠正本病的异常生化改变和改善临床症状,l即使用日间多次少量进食和夜间持续点滴高碳水化合物的治疗方案,l以维持血糖在4~5mmol/L。这种治疗措施不仅可以消除临床症状,l并且还可使患儿获得正常的生长发育。对于小婴儿可日间少量多次哺乳(母乳或以葡萄糖、i葡萄糖多聚体作为惟一碳水化合物来源的配方奶),l夜间以胃管持续滴入葡萄糖液。为避免长期鼻饲的困难,l在一岁以后可以服用生玉米淀粉混悬液,l这是近年GSD治疗最突出的进展之一,l具体剂量为1.69/kg/次,l4小时1次。随年龄增长,l剂量渐增至1.75~2.5g/kg/次,l6小时1次,l在正餐中间服用,l服用时生玉米淀粉以1:i2比例与凉白开水混合(不要开水冲服),l不宜加葡萄糖。疗效评价为餐前血糖应≥3.9mmol/L(70mg/dl),l血乳酸降至2.0~5.0mmol/L(18~45mg/dl),l夜间12小时尿乳酸应≤0.6mmol/L(5.4mg/dl)。
此外,l若患儿合并蛋白尿时应限蛋白饮食及使用ACEI类药物;用别嘌呤醇治疗血尿酸过高;低脂饮食预防高脂血症;可预防性的补充钙和维生素D;在感染期间或选择性手术前可静脉点滴葡萄糖;出现肾功能衰竭时可考虑肾移植,l肝腺瘤恶变或饮食治疗无效时可考虑肝移植。
【预后】
未经正确治疗者可因低血糖和酸中毒发作频繁而致体格和智能发育障碍。伴有高尿酸血症患者常在青春期并发痛风。成年期患者患心血管疾病、i胰腺炎和肝脏腺瘤(或腺癌)的风险明显高于正常人群,l少数患者可并发进行性肾小球硬化症。
自从应用上述饮食疗法以来,l已有不少患儿在长期治疗后获得正常的生长发育,l即使在成年后停止治疗亦不再发生低血糖等症状,l但仍需进一步追踪随访。