先天性肾上腺皮质增生症(congenital1adrenal1hyperplasia,lCAH)是一组常染色体隐性遗传性疾病,l其病因在于类固醇激素生物合成过程中某种酶的先天性缺乏,l引起肾上腺皮质合成皮质醇不足,l经下丘脑-垂体-肾上腺轴反馈调节,l促肾上腺皮质激素释放激素(corticotrophic-releasing1hormone,lCRH)、i促肾上腺皮质激素(adrenocorticotrophic1hormone,lACTH)分泌增加,l导致肾上腺皮质增生。典型的CAH发病率约为1/10000,l而非典型的发病率约为典型的10倍,l并存在种族特异性。男女发病比率为2:i1。临床主要特点为肾上腺皮质功能不全、i性腺发育异常及伴(或不伴)水盐代谢失调。
【病理生理】
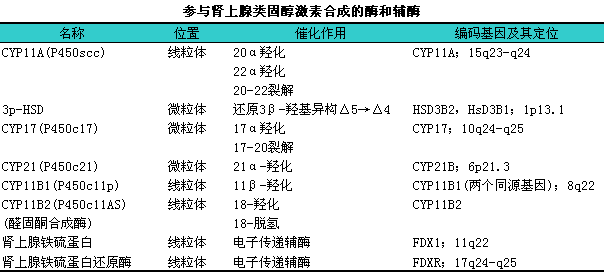
1.解剖、i生理特征11人体肾上腺由皮质和髓质两个功能不同的内分泌器官组成,l皮质分泌肾上腺皮质激素,l髓质分泌儿茶酚胺激素。肾上腺皮质又可分为3个区带:i①球状带,l位于肾上腺皮质最外层,l占皮质的5%~10%,l主要合成和分泌盐皮质激素;②束状带,l位于中间层,l约占皮质的75%,l是储存胆固醇的重要场所,l主要合成糖皮质激素,l如皮质醇及少量脱氧皮质酮(DOC)、i脱氧皮质醇(S)和皮质酮(B);③网状带,l位于肾上腺皮质最内层,l主要合成肾上腺雄激素。诸类肾上腺皮质激素均为胆固醇的衍生物,l其合成过程极为复杂,l必须经过一系列的酶促反应加工而成。在诸多类固醇激素合成酶中,l除3β羟类固醇脱氢酶(3β-HSD)外,l均为细胞色素P450(cytochrome1P450)蛋白超家族成员,l下表概括了主要参与肾上腺皮质类固醇激素合成的酶和辅酶。
2.发病机制11在正常情况下,l下丘脑分泌的CRH和垂体分泌的ACTH能促进肾上腺皮质细胞增生、i激素合成和分泌,l当血中皮质醇达到一定浓度时,l即通过反馈机制使CRH和ACTH分泌减少。若在类固醇激素合成途径中任何一个酶发生缺陷时,l都会使血中皮质醇浓度降低,l负反馈作用消失,l以致ACTH分泌增加,l刺激肾上腺皮质增生;同时酶缺陷导致前体中间代谢产物增多,l经旁路代谢可致雄激素产生过多。由于醛固酮合成和分泌在常见类型的CAH中亦大多同时受到影响,l故常引起血浆肾素活性(PRA)增高。
CAH主要包括21羟化酶缺乏症(21-OHD)、i11β羟化酶缺乏症(11β-OHD)、i3β羟类固醇脱氢酶(3β-HSD)缺乏症、i17α羟化酶缺乏症(17α-OHD)、i胆固醇碳链酶缺乏症(类脂性肾上腺增生症)等类型。其中21-OHD是最常见的CAH,l约占CAH总数的90%以上,l11β-OHD次之,l约占5%~8%,l再其次为3β-HSD缺乏症,l17α-OHD和胆固醇碳链酶缺陷症则十分罕见,l约占1%。
3.遗传特征11CAH是常染色体隐性遗传病。参与类固醇激素合成酶和辅酶的相关基因定位见下表,l均具有较高的种族差异和物种同源性。CAH的分子病理为相关基因的遗传突变,l导致编码蛋白缺陷,l故为单基因遗传病。
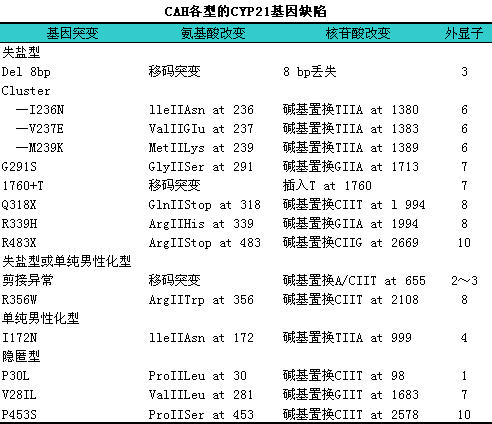
(1)CYP21B基因:i人类21羟化酶基因定位于第6号染色体短臂(6p21.3),l与HLA基因族紧密连锁。由A、iB两个基因座构成,lA基因(CYP21A)是假基因,lB基因(CYP21B)是编码21-OH的功能基因,l两者高度同源。CYP21A和CYP21B各有10个外显子及9个内含子组成,l基因全长为3463bp。1CYP21B基因突变是导致21-OHD的根本原因,l包括基因缺失、i转换和点突变等,l见表17-3。
(2)CYP11B基因:i人类编码11β羟化酶的基因为CYP11B1,l定位于第8号染色体长臂(8q21)。基因突变热点在外显子2、i6、i7和8,l至今已发现20种基因点突变。
(3)HSDB1基因:i与CAH发病相关的3β羟类固醇脱氢酶主要由HSD382基因编码表达,l与HSDB1同工酶基因的同源序列高达93%,l均定位于第1号染色体短臂(1p11-13),l由4个外显子和3个内含子组成,l基因全长约7.8kb。目前已报道的基因缺陷不少于17种,l主要包括移码突变、i无义突变和错义突变。
(4)CYP17基因:i人类17羟化酶基因定位于第10号染色体长臂(10q24-25),l包含8个外显子和7个内含子,l基因全长6.6kb。基因缺陷包括小片段缺失、i重复及点突变,l迄今未见大片段缺失报道。
类固醇激素的生物合成,l见图17-1。
【临床表现】
1.21羟化酶缺乏症(21-hydroxylase1deficiency,l21-OHD)11发病率为1/13000。临床特征为皮质醇分泌不足、i失盐及雄激素分泌过多所引起的各种表现。通常将其分为三种临床类型:i
(1)单纯男性化型(simple1virilizing,lSV):i本型约占21-OHD总数的25%,l是由于21-OH不完全缺乏所致(酶活性为正常的1%~11%)。患者不能正常合成11脱氧皮质醇、i皮质醇、i11脱氧皮质酮,l致使其相应前体物质17羟孕酮、i孕酮和脱氢异雄酮合成增多,l促使男性化表型。同时由于患儿仍有残存的21-OH活力,l能少量合成皮质醇和醛固酮,l故无失盐症状。临床主要表现为雄激素增高的症状和体征。①男孩:i同性性早熟。初生时多无任何症状,l至6月龄后逐步出现体格生长加速和性早熟,l4~5岁时更趋明显,l表现为阴茎、i阴囊增大,l出现阴毛、i变声、i痤疮等,l生长加速和肌肉发达、i骨龄提前,l但成年终身高落后,l智能发育正常;②女孩:i出生时即可出现不同程度的男性化体征:i阴蒂肥大、i不同程度的阴唇融合而类似男孩尿道下裂样改变,l子宫卵巢发育正常,l其他体格发育类似男孩。
图17-111类固醇激素的生物合成途径
(2)失盐型(salt1wasting,lSW):i本型是21-OH完全缺乏所致,l占21-OHD患者总数约75%。临床上除出现单纯男性化型表现外,l还可因醛固酮严重缺乏导致低血钠、i高血钾及血容量降低等失盐症状的出现,l表现为呕吐、i腹泻、i脱水、i消瘦、i呼吸困难和紫绀等。常因诊断延误、i治疗不及时在出生2周内死亡。
(3)非典型型(nonclassic,lNC):i亦称迟发型或轻型,l是21-OH轻微缺乏所引致的一种变异型。症状轻微,l临床表现各异。发病年龄不一,l多在肾上腺功能初现年龄阶段出现症状。男孩为阴毛早现、i性早熟,l生长加速、i骨龄超前;女孩表现为初潮延迟、i原发性闭经、i多毛症、i不孕症等。
2.11β羟化酶缺乏症(11β-hydroxylase1deficiency,l11β-OHD)11发病率为1/10000。临床可分为典型与非典型型。因11β-OH缺乏而导致DOC增加,l可使部分患儿出现高血钠、i低血钾、i碱中毒及高血容量,l故有2/3患者出现高血压症状;又因皮质醇合成减少引起肾上腺雄激素水平增高,l出现类似21羟化酶缺乏的高雄激素症状和体征。但一般女孩男性化体征较轻,l男孩出生后外生殖器多正常,l至儿童期方出现性早熟体征。非典型型临床表现差异较大,l部分患儿可至青春发育期因多毛、i痤疮和月经不规则而就诊,l大多血压正常,l男孩有时仅表现为生长加速和阴毛早现,l临床较难与21-OHD的非典型患者区别。
3.3β羟类固醇脱氢酶(3β-hydroxysteroid1dehydrogenase,l3β-HSD)缺乏症11典型病例出生后即出现失盐和肾上腺皮质功能不全的症状,l如厌食、i呕吐、i脱水、i低血钠、i高血钾及酸中毒等,l严重者因循环衰竭而死亡。男性可有不同程度的外生殖器发育不良,l女性则出现不同程度男性化。非典型病例约占本症10%~15%,l出生时往往无异常,l至青春发育期前后出现轻度雄激素增高体征,l如女孩阴毛早现、i多毛、i痤疮、i月经量少及多囊卵巢等。
4.17羟化酶缺乏症(17-hydroxylase1deficiency,l17-OHD)11由于皮质醇和性激素合成受阻,l而DOC和皮质酮分泌增多,l导致临床发生低钾性碱中毒和高血压,l女性青春期呈幼稚型性征和原发性闭经;男性则表现男性假两性畸形。
【辅助检查】
1.生化检测11尿液包括17羟类固醇(17-OHCS)、i17-酮类固醇(17-KS)和孕三醇;血液包括钠(Na)、i钾(K)、i肾素血管紧张素原(PRA)、i醛固酮(Aldo)、i17羟孕酮(17-OHP)、i脱氢异雄酮(DHEA)、i脱氧皮质酮(DOC)及睾酮。临床检测判断意义参见下表。

2.基因分析
(1)直接聚合酶链反应(PCR):i应用该项技术可直接检测相关基因缺失。
(2)聚合酶链反应-寡核苷酸杂交(PCR-ASO):i将PCR扩增产物与特异性寡核苷酸探针(野生型和突变型)杂交,l并根据杂交条带的特异性鉴别相关基因突变性质。
(3)聚合酶链反应-限制性内切酶片段长度多态性(PCR-RFLP):i利用基因点突变可造成限制性内切酶的酶切位点改变(产生或消失酶切位点),l将PCR扩增产物进行相应酶解反应,l从中判断是否存在相应的基因突变,l如CYP21B基因突变(见表17-3)。
【诊断和鉴别诊断】
本病应争取早期诊断和及时治疗(见下表)。新生儿期失盐型患儿应与幽门狭窄、i食道闭锁等症相鉴别;儿童期患儿应与性早熟、i真两性畸形、i男(或女)性化肾上腺皮质肿瘤、i性腺肿瘤等相鉴别;

【治疗】
治疗原则:i①一经诊断应立即早期给予治疗;②首选肾上腺皮质激素类药物;③药物剂量因人而异;④应激情况应加大剂量;⑤女性患者及失盐型男女患者应终生治疗,l单纯男性化型的男性患者在进入青春期和成年期后可酌情停药。
1.失盐型患儿必须及时纠正水、i电解质紊乱11可用生理盐水或0.45%盐水及碳酸氢钠溶液进行补液,l但不能使用含钾溶液。必要时可肌肉注射醋酸脱氧皮质醇(DOCA)1~3mg/日,l或口服氟氢可的松(9α-fludrocortisone)0.05~0.1mg/日,l剂量应按当日补给的氯化钠量适当调整。脱水纠正后将糖皮质激素改为口服,l并长期维持,l同时给予氯化钠2~4g/日。
2.糖皮质激素的使用11大多应用氢化考的松,l按每日10~20mg/m2计算,l2/3量晚间用,l1/3量分次白天服用。该类药物可替代肾上腺皮质醇的分泌不足,l并抑制过多的ACTH合成,l从而减少过多产生雄激素,l达到改善男性化等症状的目的。
3.盐皮质激素的应用1121羟化酶缺乏症患儿无论是否失盐,l其血浆肾素活性都很活跃,l应用氟氢可的松可协同糖皮质激素作用,l使ACTH分泌进一步减少。一般口服氟氢可的松的剂量是0.05~0.1mg/日,l待症状改善后可酌情减量。应注意0.1mg氟氢可的松相当于1.5mg氢化可的松,l在使用时应扣除相当的皮质醇用量,l以防皮质醇过量。
在皮质激素治疗过程中必须进行临床评估及监测,l包括血浆17-OHP、iDHEA、iT、iPRA、i电解质及尿17-酮的测定,l以调节两类激素的用量,l达到最佳治疗效果。
4.外科治疗11在药物控制前提下可行外阴矫治术。
【预防】
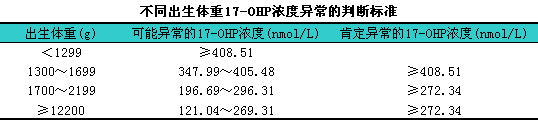
1.新生儿筛查11主要是新生儿21羟化酶缺乏症的筛查。目的是:i①预防危及生命的肾上腺皮质危象及盐皮质功能不足而导致的CAH患儿脑损伤或死亡;②预防女性患儿由于外生殖器男性化造成性别判断错误;③预防过多雄激素造成患儿日后身材矮小、i心理生理发育等障碍。方法:i生后2~5天足跟采血滴于特制滤纸片上,l经ELISA、i荧光免疫等方法测定17-OHP浓度来早期诊断。正常新生儿出生后17-OHP可增高,l至12~24小时后降至正常。判断标准参见下表。
2.产前诊断11因CAH是常染色体隐性遗传病,l每生育一胎就有1/4概率为CAH患者。因此,l对家族中有本病先证者的孕妇应做产前诊断,l并及时给孕妇地塞米松进行胎儿预防性治疗。①21-OHD通常在孕9~11周取绒毛膜(CVS)活检进行胎儿细胞DNA分析,l孕16~20周取羊水(AF)检测孕三醇、i17-OHP等生化项目。由于大部分非典型21-OHD患儿出生后17-OHP水平未明显升高,l因而无法通过新生儿筛查而发现,l基因检测是此型患儿唯一早期诊断的手段。②11β-OHD产前诊断类似21-OHD,l主要测定羊水DOC及取绒毛膜进行相关基因分析。