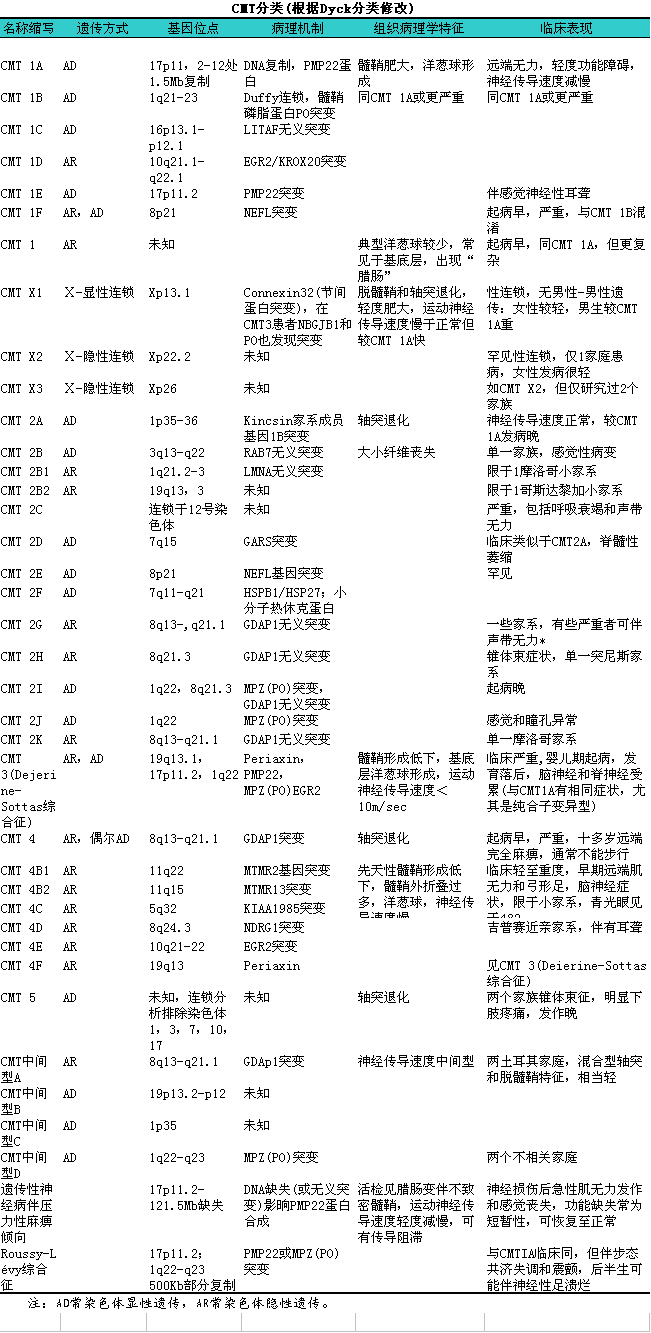
遗传性运动感觉神经病2型(CMT2)(表8—54)
欧洲神经肌肉中心111997年1.临床诊断标准
(1)家族史
1)肯定诊断:i常染色体显性、i隐性或X-连锁遗传。
2)备注:i单独病例或为隐性遗传或为显性遗传性新突变的结果。
(2)起病年龄
肯定诊断:起病年龄变化大,l常在20岁以内。
(3)肌萎缩和无力:i
1)肯定诊断:i下肢远端突出的肌萎缩和无力,l常为对称性。足和足趾的伸肌、i足内肌群较小腿肌受累更早更严重。
2)备注:i以后,l手内肌群、i股内侧肌的远端、i股四头肌的其他部分也可出现萎缩和无力。手肌无力很少作为首发症状。部分患者,l尤其是常染色体隐件遗传病例可出现肩部肌萎缩。可能出现小腿肌肥大。
(4)其他相关特征
1)备注:i感觉受损常见但非必须具备的特征。弓形足,l脊柱侧凸,l震颇、i溃疡.感觉神经性耳聋,l智力迟缓。横膈肌和声带麻痹。
2)排除诊断:i主要表现为中枢神经系统包括锥体束或小脑体征。
(5)病程和严重度
肯定诊断;缓慢进展,l严重度不同。
2.实验室诊断标准
(1)代谢方面:i
排除诊断:i神经病的代谢性和获得性原因。肌酸磷酸激酶明显升高。
(2)电生理。
1)肯定:i家族中至少有一人正中神经神经传导速度>38m/s。感觉神经动作电位减小或消失。肌电图呈现神经源性变。
2)备注:i某些家族成员正中神经神经传导速度<38m/s不影响CMT2诊断。常有肌电图失神经性变的证据。复合肌肉动作电位振幅降低常见。病程进展的患者单根神经(常为腓神经)运动神经传导速度不能测得。
(3)感觉神经活检:i
1)肯定诊断:i大的有髓鞘纤维密度和总数降低。成簇的薄髓鞘的退化纤维。
2)备注:i大的有髓鞘纤维横向神经丛区域明显减少。可能偶尔出现小洋葱球。
(4)分子遗传学:i
1)排除诊断:i排除CMT1A型的复制。
2)备注:i已识别CMT2位点在染色体lp35-p36(CMT2A),l3q22-q23(CMT2B)和lp14(CMT2D)上。
远端型遗传性运动神经病(远端型HMN)或脊髓CMT病诊断标准。
1.临床诊断标准
(1)家族史:i
1)肯定诊断:i常染色体显性或隐性遗传。
2)备注:i单独病例或为隐性遗传性远端HMN或为显性遗传性新突变的结果。
(2)起病年龄:i
1)肯定诊断:i常在10岁以内。
2)注释:i起病年龄变化大。先天性至晚发性。
(3)肌萎缩和无力:i
1)肯定诊断:i肢体远端为主的肌萎缩和无力。
2)备注:i肌无力和萎缩通常为下肢远端,l最初在腓肠肌和足内肌群。某些家庭,l先于足背屈肌无力的小腿肌无力和萎缩可为最早的体征。很少一部分病人,l手和前臂肌无力为起病症状。可为不对称性发病。以下肢远端肌无力起病的病例;在疾病最后阶段可发生手内肌和下肢远端肌肉萎缩和无力。可出现小腿肌肉肥大。
3)排除诊断:i肌无力以肢体近端为主。
(4)其他相关特征:i
1)备注:i关节弯曲、i弓形足、i脊柱侧凸、i震颤、i进行感觉神经性耳聋、i声带麻痹。
2)排除诊断:i感觉受损。中枢神经系统包括锥体束明显受累。
(5)病程和严重度:i
肯定诊断:i缓慢进展,l严重度不同。
2.实验室诊断标准
(1)代谢方面:i
排除诊断:i神经病的代谢性和获得性原因。肌酸磷酸激酶明显升高。
(2)电生理:i
1)肯定诊断:i运动神经传导速度正常或轻度减慢.感觉神经传导速度正常。肌电图呈现神经源性变。
2)备注:i病程进展的患者单根神经(常为腓神经)运动神经传导速度不能测得。
3)排除诊断:i远端肌肉肌病性异常。
4)备注:i除年龄相关性改变外,l腓肠神经活检正常。
(3)分子遗传:i
排除诊断:i与染色体5q11.2-13.3运动神经存活基因突变相关的近端性脊髓性肌肉萎缩。