伴皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病
一、i疾病概述
伴皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病(cerebral1autosomal1dominant1arteriopathy1with1subcortical1in1farcts1and1leukoencePhalopathy,lCADASIL)是19p13上的Notch-3基因突变引起的显性遗传的非动脉硬化性、i非淀粉样变的脑小动脉病。大多数在中年早期起病,l呈常染色体显性遗传;以反复脑缺血性发作,l渐进性认知障碍、i痴呆为主要临床特征;可伴有先兆型偏头痛和(或)情感障碍。影像学上表现为皮质下白质脑病和多发腔隙性梗死,l以磁共振成像(MRI)上的颞叶前部T2加权像高信号为特征。电镜下可见脑或全身小动脉中膜内平滑肌细胞表面有特异性的嗜锇颗粒物质(GOM)沉积。
【病因与流行病学】
目前认为该病是单基因遗传的缺血性脑血管疾病,l致病基因为19号染色体短臂上的Notch-3基因。1993年Tournier-Lasserve等首先将该病的致病基因定位于19号染色体,l而Sabbadini等进一步将致病基因定位于19p13.2~19p13.1上的Noteh-3基因,l并证实Notch-3基因的外显子(最常见的足第4号外显子,l其次为第3、i5、i6号外显子)发生突变。首届国际CADASIL研讨会(1993)在法国巴黎共识并统一命名了该病。该病的发病率未见报道。发病年龄在30岁~40岁之间,l卒中症状出现的平均年龄为45岁,l50岁以后逐渐出现认知障碍和痴呆。发病年龄在性别上无差异。最常见的症状是卒中和痴呆,l同家族不同代之间无差别。病程10年~30年,l平均死亡年龄65岁。本病并非罕见,l欧洲、i美洲、i亚洲、i非洲、i澳洲均有报道。随着对该病认识的提高和诊断技术的改进,l全球该病的病例数正处于上升阶段。
【发病机制】
尚不完全清楚。迄今已知Notch-3基因突变有50多种.大多数是错义突变。80%为碱基C突变为T,l使得氨基酸序列中的精氨酸被半胱氨酸所替代。Notch-3基因的突变改变了蛋白结构,l干扰了配体一受体的相互作用,l导致血管内皮细胞的构造异常,l最终引起血管平滑肌细胞损害。致使脑内外的小动脉中膜增厚和颗粒样嗜碱性物质沉积,l电镜下可见局灶性血管平滑肌破坏和GOM在其表面的沉积。这种改变导致了以额、i颞、i顶、i枕叶为主的病变.在大脑皮质下出现白质弥漫性或局灶性脱髓鞘,l在脑深部的白质、i丘脑和基底核等处出现多发腔隙性梗死。
该病脑组织的典型病理改变是以皮质下白质缺血性脱髓鞘、i多发腔隙性梗死和脑小动脉特异性改变为特征。病变主要累及200μm~400μm的微小动脉。光镜下可见病变部位的小动脉内膜下纤维增生和玻璃样变性,l管壁向心性增厚与管腔狭窄。小动脉中膜出现颗粒样PAS阳性物质;电镜下显示脑或软脑膜小动脉的平滑肌细胞表面有特异的直径约10nm~15nm的GOM沉积。
【临床表现】
CADASIL主要有四大临床表现:i脑缺血性发作、i认知功能障碍或痴呆、i先兆型偏头痛和(或)情感障碍。
1.脑缺血性发作(包括TIA或皮质下缺血性卒中)为中年期的主要表现,l85%为短暂性脑缺血发作或缺血性卒中。该病的重要特点之一是约有84%患者没有高血压、i糖尿病、i高血脂等脑血管病危险因素。各型皮质下缺血(包括短暂性脑缺血发作、i可逆性缺血性神经功能缺失和完全性卒中)均可出现,l约2/3为典型的腔隙性梗死综合征:i纯运动性卒中、i共济失调轻偏瘫、i构音障碍手笨、i纯感觉性卒中和感觉运动性卒中。在整个病程期间,lTIA的次数为1次~10次,l缺血性卒中发作次数大多数在2次~5次之间。
2.认知障碍或痴呆是中、i老年期的主要表现。认知障碍确切起病时间较难确定。90%认知障碍逐渐发生,l逐步恶化,l属皮层下型。以额叶功能受损为主,l表现为记忆、i注意障碍,l执行和视空间功能障碍以及情感淡漠等。痴呆出现的年龄为40岁~70岁,l65岁以上者80%有痴呆。与痴呆伴随的症状有步态异常(90%),l尿失禁(86%)和假性球麻痹(52%)。痴呆的发生与脑缺血发作均具有显著相关性,l有痴呆患者比无痴呆患者更容易发生缺血性卒中。基底核和丘脑部位的缺血性损害对CADASIL患者早期发生痴呆有重要的影响。一般失语、i失用或失认罕见,l只有在基底核病变极其严重时才出现皮质代谢降低,l提示该病的痴呆发生与基底核梗死的远隔效应有关。
3.先兆型偏头痛11平均发病年龄25岁,l约占20%~30%,l是青年期的主要表现。有先兆症状的偏头痛发作前可有5min~15min单侧或双侧视觉模糊或感觉异常等,l一般不超过1h。典型偏头痛发作常呈单侧剧烈头痛伴有血管搏动感、i恶心、i呕吐、i畏光、i怕声等症状。多数患者反复发作,l每次持续时同为2h~48h,l发作间期可无症状。也有个别患者表现为偏瘫型偏头痛、i基底型偏头痛或仪有先兆症状,l不易与脑缺血发作区别。
4.情感障碍 情感或心境障碍是该病最常见的精神症状,l约占20%~30%。多数表现为严重抑郁障碍,l少数可与典型的躁狂发作交替出现。有早期诊断为双向性情感障碍,l直至MRI检查后才发觉有误。此外,l偶见惊恐、i幻觉和妄想发作。发生情感障碍的确切原因仍未明,l而基底核和额叶白质缺血的部位可能在其发生中起关键作用。其他少见的表现包括癫痫发作(10%)、i锥体外系症状、i耳聋等。一般不累及颅神经、i脊髓或肌肉组织。
【诊断和鉴别诊断】
1.诊断标准目前国内外尚无统一诊断标准,l鉴于该病的临床表现复杂多样,l认为只有结合病史(包括发病年龄、i家族遗传史、i有无高血压等血管病危险因素)、i神经影像学检查、i皮肤血管活检和基因检测才能做出正确的诊断。
Marcus等(2002)比较了各种诊断方法的特异性和敏感性,l制定了如下诊断标准:i①中年起病,l有明确的家族史,l80%的患者无高血压、i淀粉样变及其他血管病危险因素;②有缺血发作,l有先兆型偏头痛,l认知功能障碍和情感障碍等表现中的一项或多项;③脑MRI检查在T2加权像和FLAIR像上可见大脑深部和基底节区呈现对称性的白质高信号,l常伴腔隙性梗死灶.绝大多数有中至重度颞叶前部和外囊受累;④皮肤、i肌肉活检:i在电镜下可见特异性嗜锇颗粒物质(GOM)沉积;⑤基因测序可发现Notch-3基因突变。
2.鉴别诊断
(1)Binswanger病:i此病的反复发作性脑卒中,l皮质下痴呆,l缺血性白质脑病与CADASIL相似。但此病起病年龄较晚,l常有高血压、i糖尿病等脑血管病危险因素,l无先兆型偏头痛,l无家族遗传史,l缺乏特征性的颞极自质T2高信号,l缺乏小动脉超微结构的特征性改变,lNotch-3基因突变有助于鉴别。
(2)多发性硬化(MS):i此病主要累及皮质下白质,l其临床和影像学表现与CADASIL相似,l但多为散发,l常表现CADASIL所没有的视神经、i脊髓和小脑病变,l鞘内合成的IgG增高,l无脑缺血发作和O’Sullivin征有助于区别。
(3)颅内原发性血管炎:i此病往往散发,l呈现双侧多发性幕上病变,l常累及大脑皮质。以炎性病理改变为主,l不同于CADASIL。
(4)脑底异常血管网症:i又称烟雾病,l以其颈内动脉终末段的严重狭窄或闭塞,l以及影像学上特征性的烟雾样改变不难与CADASIL区别。
(5)伴视网膜病肾病和卒中的遗传性血管内皮细胞病(HERNS):i此病也累及全身小血管,l也呈常染色体显性遗传,l也在中年出现卒中发作,l常伴精神症状或偏头痛,l酷似CADASIL。但此病常伴后者所没有的视觉(视野暗点)和肾脏损害(肾功能不全和蛋白尿),l病理上以血管内皮细胞的基底膜显著增厚为特征,l而无小动脉中膜平滑肌细胞表面的GOM沉积。
(6)线粒体脑肌病伴高乳酸血症和卒中样发作综合征(MEALAS):i40岁前起病,l儿童期更多。以反复卒中样和癫痫样发作为特征,l头颅CT和MRI上显示枕叶脑软化,l而无颞极白质T2高信号,l病灶范围与主要脑血管分布不一致(层状坏死);血和脑脊液乳酸增高。肌肉活检可见特征性的破碎红纤维(RRF),l电镜下在肌细胞内可见到线粒体堆积。不难与CADASIL鉴别。
此外,l该病还要注意与其他遗传性血栓性疾病如高同型半胱氨酸血症(HCY)、iFabry病、i因子V1Leiden突变鉴别;与其他遗传性疾病如遗传性异常脂蛋白血症、i遗传性淀粉样血管病、i肾上腺脑白质营养不良、i显性遗传性正染性白质脑病等相鉴别。
总之,l在临床鉴别有困难时均可求助于超微结构和分子遗传学检查,l上述疾病均未见血管平滑肌细胞表面出现GOM沉积,l亦无Notch-3基因突变。
二、i检验诊断
CADASIL目前实验室常规检查项目较少,l其实验室诊断主要依据分子生物学技术如Notch-3基因突变检查以及病理柱查。
【一般检验项目】
1.脑脊液细胞计数及分类
(1)检测方法:i显微镜镜检法。
(2)标本:i脑脊液、i脑池液、i脑室液。
(3)参考范围:i无色透明,l白细胞0~5×106/L,l以单核细胞为主,l红细胞为0。
(4)临床诊断价值和评价:i大多数病例无色透明,l尤其是24h后仍无色透明则支持缺血性脑血管病,lCADASIL病也不例外。细胞数大多正常,l但出现梗死时,l白细胞增多,l通常小于50×106/L,l如果梗死继发出血.CSF中可出现红细胞,l甚至血性(红细胞在11000×106/L)。
(5)方法学评价和问题:i见其他相关章节。
2.脑脊液总蛋白
(1)检测方法:i双缩脲干化学法、i磺柳酸硫酸钠比浊法。
(2)标本:i脑脊液、i脑池液、i脑室液。
(3)参考范围:i蛋白定性(Pandy试验)阴性,l定量试验蛛网膜下腔CSF为0.15g/L~0.5g/L,l脑池液为0.1g/L~0.25g/L,l脑室液为0.05g/L~0.15g/L。
(4)临床诊断价值和评价:iCADASIL患者的脑脊液总蛋白一般会增高,l尤其是在皮质下梗死发作时,l但一般不超过1g/L。
(5)方法学评价和问题:i双缩脲干化学法具有快速、i准确、i重复性较好的特点,l适合急诊检验;磺柳酸_硫酸钠比浊法为早期蛋白定量方法,l该法线性范围窄,l重复性差,l影响因素较多,l一般适合门诊或基层单位使用。
3.同型半胱氨酸(HCY)
(1)检测方法:i酶免疫化学发光法。
(2)标本:iEDTA抗凝血血浆、i脑脊液。
(3)参考范围:i≤15μmol/L。
(4)临床诊断价值和评价:i有学者研究发现,lCADASIL患者的HCY水平明显高于正常对照组,l说明高同型半胱氨酸血症与CADASIL的发病病程有一定的关系。
(5)方法学评价和问题:i见其他相关章节。
【特殊检验项目】
1.病理学分析
(1)检测方法:i显微镜检。
(2)标本:i病变组织。
(3)临床诊断价值和评价:iCADASlL病理改变为脑深部多发的腔隙梗死和白质脑病,l以及小动脉中层平滑肌细胞表面GOM的沉积,l这些特征性的病理改变是渗断CADASIL的金标准。
2.1Notch-3基因突变
(1)检测方法
1)盐析法提取患者外周血基因组DNA,l酚/氯仿法提取肌肉组织基因组DNA。
2)聚合酶链反应(PCR):i①引物覆盖了Notch-3基因编码区的外显子1~12及其两旁的内含子序列;②25μl1PCR反应体系中含引物各0.2μmol/L,ldNTP1100/μmol/L,lTris-HCl10mmol/L,lKCl150mmol/L,lMg2+1.5mmol/L,l0.1%Tri-tonX-100,lTaq酶1U,lDNA模板50ng~100ng;③PCR反应条件:i94℃预变性5min,l94℃变性45s,l56℃退火45s,l72℃延伸45s,l共30个循环,l最后1个循环于72℃延伸10min;④反应结束后,l以1.5%琼脂糖凝胶(内含0.5μg/ml的溴化乙啶)电泳检测PCR产物。
3)取100μl1PCR产物进行扩增、i纯化,l然后用DNA全自动测序仪测序,l对发现的突变再进行反向测序证实。
(2)标本:i2mlEDTA-K2静脉血、i脑活检组织。
(3)临床诊断价值和评价
1)有学者利用正、i反向测序证实先证者存在Notch-3基因的杂合性突变,l且突变位点各不相同,l有的突变位于外显子3,l分别为268C→T、i322C→T和328C→T突变(核苷酸的位置从翻译起始密码子ATG的A算起),l造成Notch-3蛋白质突变分别为R90C、iC108R和R110C;有的先证者突变位于外显子11为1819C→T.造成R607C。其他常见基因的错义突变见下表。
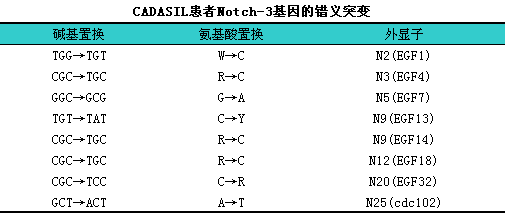
2)CADASIL是由Notch-3基因突变引起,lNotch-3基因突变所致的血管平滑肌细胞的变性可能在CADASIL的发病机制中起重要作用。
(4)方法学评价和问题
1)基因检测(即DNA测序)可以十分有效地渗断该病,l但若基因突变的部位不发生在最常见的第4号外显子或较常见的第3、i5、i6号外显子时,l测序十分耗时,l同时因外显子数量多而使检测费用昂贵。
2)病理改变为脑深部多发的腔隙梗死和白质脑病,l以及小动脉中层平滑肌细胞表面GOM的沉积,l这些特征性的病理改变是诊断CADASIL的金标准。
3)该病PCR方法检测成功的关键在于引物的设计,l建议用primer软件设计引物。CADASIL诊断用的常用引物见下表。

【应用建议】
1疑CADASlL时,l首选检验组合建议:i病理学分析+同型半胱氨酸(HCY)+脑脊液细胞计数及分类。
2.CADASIL的分子生物学诊断检验组合建议:i病理学分析+Notch-3基因突变检测。