一、i疾病概述
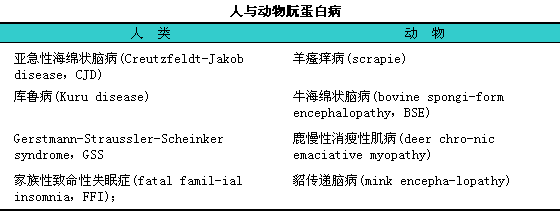
朊蛋白病是一组由变异朊蛋白引起的可传染性、i慢性进展性、i致死性海绵状脑病,l朊蛋白病最早发现于动物,l200年后才发现其可在人类发生。已明确发生在人类和动物的朊蛋白病各有4种(见下表)。
【病原学】
本病的病原体为一种有传染性的蛋白质(pro-teinacious1infectious1particles),l即prion,l中译名为朊蛋白或感染性蛋白,l简称PrP。虽然有些学者认为它可能含极微量的核酸(<50个核苷酸),l但PrP是否含有核酸尚无定论,l因而不属病毒范围。它能耐高热、i核酸酶、i紫外线等的分解作用,l但多种蛋白酶、i蛋白变性剂、i去污剂、i有机溶媒和尿素等能降低其传染性,l说明蛋白质是引起感染的核心成分。
其核心为分子量27~30kD蛋白,l称为PrP27~30。这个耐热蛋白酶的核心的外面覆盖着一层分子量为33~35kD的蛋白,l称为PrP33~35或PrPSC。另一个与PrPSC相似的蛋白PrPC1见于未感染的正常动物脑中,l它能耐受蛋白酶的消化,l并可聚合成被称为PrP杆的杆状结构或羊瘙痒病相关纤丝(SAF)。根据PrP基因的序列分析,l发现它是由宿主基因所编码而不是病原体的一部分,l该基因位于人第20号染色体的短臂上,l其可阅读框架由1个外显子组成,l系253个氨基酸的围绕着PrP33-35的蛋白。未发现在正常和羊瘙痒病感染动物PrP的结构之间有任何区别。在细胞培养物以及脑脊液中可检出分泌型PrPC,l相反PrPSC则积聚于细胞内的空泡和溶酶体,l而不见于细胞表面。关于PrPSC的积聚如何引起神经元功能紊
乱的机制还不清楚,l大鼠海马回细胞培养暴露于围绕着PrP的第106~126氨基酸后发生细胞凋亡的事实提示PrP或其衍生物可能有神经毒性。
【流行病学】
本病为全球性发病,l在美、i欧、i澳、i亚等洲人口密集地区,l发现1年的发病率(1~3)/10万,l全球CJD罹患率仅为百万分之一。Kahana(1994)报道以色列各种族的年发病率为(0.4~1.9)/10万。死亡率略同于发病率,l从发病到死亡的平均时期约为1年。发病年龄为35岁~71岁,l高峰为55岁,l35岁以下罕见,l女性较男性多,l男:i女=2:i3。其中85%~95%为散发型,l散发型发病年龄为55岁~70岁,lnvCJD发病年龄为19岁~39岁。未发现有人与人之间的传播。曾报告24例医务人员中发生孤立的CJD,l但其发病率不超过单纯由于机遇所致。未发现有患者向医务人员或殡葬从业人员的传播。家庭内多个病例的发生纯粹由于遗传因素而与接触传播无关。
由于基因的变异、i密码子的置换使朊蛋白病的临床表现变化多样,l复杂纷呈。现以最常见的、i临床研究相对清楚的亚急性海绵状脑病为代表,l做一简述。
(一)亚急性海绵状脑病
亚急性海绵状脑病是一组由变异朊蛋白引起的海绵状脑病,l以精神障碍、i痴呆、i帕金森样综合征、i共济失调、i肌阵挛、i肌肉萎缩为特点的可传染性、i慢性进展性、i致死性疾病。最早由Creutzfeldt和Jakob分别于1920年和1921年描述,l故以他们的名字命名该病Creutzfeldt-Jakob1disease,l以下简称CJD。本病共分四型:i①散发型;②家族型;③意外感染型或获得型;④变异型(nvCJD)。
【病因和发病机制】
1.散发型CJD其病因不明。
2.家族型CJD与PrP基因突变相关。约5%~15%病例有家族史,l呈典型的常染色体显性遗传。家族型在某些国家中较为多见,l主要为利比亚、i北非洲和斯洛伐克。
3.意外感染型或获得型11与暴露于CJD患者组织相关,l如角膜移植、i硬脑膜移植和垂体功能低下患者多次接受从尸体脑垂体制备的生长激素治疗而受感染。
4.变异型CJD(nvCJD)11与食用BSE污染的牛制品相关。1986年在英国发现牛海绵状脑病(BSE),l又名疯牛病,l它是通过以受感染动物尸体加工成饲料喂养动物而传播的。1996年3月在英国发现的10例nvCJD,l虽然其发病年龄较小,l病理改变及临床表现和经典型的qD均有所不同,l用免疫印迹法证明引起BSE的PrPSC和引起10例nvCJD的PrPSC显示出同一个电泳模式,l强烈提示BSE和nvCJD是由同一个病原体所引起的。
【病理改变】
本病是一种大脑、i小脑、i丘脑和脊髓灰质均受累的疾病。尸体解剖可见脑萎缩。组织病理学改变以大脑灰质及纹状体最为严重,l但小脑和丘脑的病变也可明显,l不影响脑白质。显微镜下呈现广泛的海绵状态,l神经细胞丧失和明显的胶质细胞增生,l神经纤维网(即轴突、i树突和胶质纤维)内出现小圆形空泡,l无明显脑膜反应和炎性变化。概括为三种:i①海绵样变性:i不是持久可见到的改变,l特别是病程长的患者,l此改变显著时,l皮质的一些区域可能被包绕以星形细胞和少数神经元残体的蜂窝状小囊所取代;②神经元变性:i见于大脑皮质、i基底节、i丘脑、i小脑、i脑干和脊髓的前角细胞,l不同的患者病变轻重不等。病变为双侧性,l但常不对称,l有些患者重点损害额叶和丘脑,l有些患者则是损害枕叶和小脑,l脊髓町严重受损,l但也有不受波及者;③星形细胞增殖常较神经元的丧失广泛,l似不象仅是由于细胞变性的反应所引起。此外,l还可见到淀粉样斑块(类淀粉纤维斑,lKuru斑),l这也是病理变化的一种特征性改变。特别是当病变主要损害小脑时,l出现率明显提高。
超微结构研究神经元的树突和神经胶质细胞内可显示空泡,l但空泡不是空的,l而是含有与细胞膜碎片相似的卷曲的结构。其外观与羊瘙痒病有关的纤丝(SAF)类似,l这些纤丝的确切成分尚不清楚。界膜空泡样变性,l导致独立膜单位破坏,l引起细胞问融合,l改变了神经元兴奋性,l形成了成组电位发放,l从而在临床上出现肌阵挛发作。
免疫组织化学方法诊断人类朊病毒病是重要进展,l用抗PrP血清检测人朊病毒是一项敏感的检测手段。
【临床表现】
本病各年龄段均可发病,l但50岁~70岁最多见。男女发病率大致相等。基本特点是起病隐袭,l潜伏期3年~22年,l进行性加重,l局限于中枢神经系统多见。根据病程进展情况可分为两组:i进展缓慢组和进展迅速组。
1.进展较迅速组约占80%,l呈亚急性经过,l从起病至死亡的平均病程为7个~9个月。本病的临床症状和体征较复杂,l主要为皮层功能障碍和运动失调,l特点是迅速进行性痴呆和肌阵挛发作。出现率分别为98%~100%和92%。此型CJD的临床可分三个阶段(三期)。
(1)第一阶段(前驱期):i数周或数月,l大约1/3患者有前驱症状。记忆减退、i失眠、i头痛、i头昏、i心理变化、i性格改变、i肢体无力、i精神异常(多为非特异性的情感改变,l以忧郁为多),l可迅速衰退。
(2)第二阶段(痴呆期):i除进行性痴呆、i肌阵挛外可有锥体系、i锥体外系、i小脑、i下运动神经元、i视力障碍等受损的症状,l根据临床表现不一致可划分为6种临床亚型:i
1)额顶锥体柬型海绵状脑病:i即所谓Jakob痉挛性假性硬化型。主要表现为进行性痴呆、i肌强直、i肌阵挛发作和锥体束征等;可有肌阵挛,l迅速地呈刻板跳跃性运动,l于一个部位持续达数分钟,l然后又转至其他部位。
2)枕颞视力障碍型海绵状脑病:i即Heidenhain综合征。视力障碍明显,l包括视野缩小,l皮质性失明和视觉性认识不能,l未观察到视乳头水肿。伴有进行性痴呆和其他体征。
3)共济失调型海绵状脑病:i明显的小脑性共济失调,l伴痴呆和肌阵挛等表现;疾病早期明显,l当肢体出现严重的强直或痉挛时,l就不易查及共济失调。
4)肌萎缩型海绵状脑病:i病变累及皮层和明显累及延髓和脊髓前角细胞,l临床表现为痴呆和突出的延髓、i脊髓症状。
5)全脑或弥散型海绵状脑病:i病变广泛累及中枢神经系统,l临床表现为进行性痴呆和肌阵挛发作、i锥体束征、i锥体外系征和小脑体征;最终表现为运动机能减退、i肌强直、i小脑症状、i眼球震颤和共济失调。
6)其他变异型海绵状脑病:i包括家族性老年性痴呆等。
(3)第三阶段(临终期):i患者常处于昏迷、i木僵或去大脑强直状态,l也可呈植物状态,l最终因肺部感染加重而死亡。
2.进展较缓慢组在病程较长的20%患者中,l病期为20个~57个月。初发症状在某些方面有所不同,l在出现明显的其他器质性疾患以前几个月,l多有进行性痴呆,l除疾病进展相对缓慢外与病程较急性发展组还存在显著差别,l多数患者在病程早期存在广泛的肌肉萎缩,l乃由于前角细胞损害所致,l相反,l肌阵挛和视力丧失在本组的患者中罕见。
CJD患者具有特征性的脑电图改变,l脑电图对其的诊断具有重要价值。典型脑电图改变可见于75%~90%病例,l早期仅示轻微、i过多的普遍性慢渡;随着病程的发展,l可见有特色的间歇期为0.5秒~1.0秒重复出现的尖渡(PSD)或在慢波背景上出现普遍双侧的同步双相或三相周期性尖渡复合波(PSWCS),l间隔为0.5秒~2.5秒,l持续200毫秒~600毫秒.也可称为周期性变化模型(CAP),l是诊断CJD的脑电图指征。PSD的脑电图变化模式可肯定CJD之诊断,l总出现率为60%~70%,l在病程的三个阶段出现率分别为33%、i94%及78%,l半单侧亦可见。国内报道72.3%CJD患者出现PSD,l在发病10周后出现,l与国外学者关于发病10周内无PSD模式即否定CJD的观点不同。脑电图有PSD可肯定诊断,l无则不能轻易放弃和排除诊断,l更不能因10周内脑电图不出现PSD而否定诊断。定期复查EEG有诊断意义,l如连续4个月未检出特征性EEG改变时,l应怀疑CJD诊断。不同临床类型、i病程、i病灶部位、i范围大小、i起病方式以及脑电图描记次数和时间长短均会影响脑电图结果。要注意,lCJD的脑电图异常慢波亦有报告占64%,l即有从发病到死前脑电图均正常者。
单纯的CT检查无特异性意义,l但迅速进行性智力丧失而不伴有显著脑萎缩时则提示有CJD的可能,l因为Alzheimer病的典型改变为显著脑萎缩。MRI检查比CT更为敏感,l有皮层萎缩而不伴自质改变。PET可证实两侧半球不对称的颞叶代谢降低。
【诊断】
1.散发性CJD诊断标准(WHO)
(1)很可能:i①进行性痴呆;②典型的脑电图表现,l即每秒出现的典型三相波或14-3-3蛋白阳性;③至少加上以下两项条件:i肌阵挛、i视觉或小脑症状、i锥体或锥体外症状、i无动性缄默。
(2)可能:i临床症状和体征与上述相同,l但无脑电图的特殊改变,l且病程在1年以上。
2.意外感染CJD诊断标准(WHO)
(1)接受脑垂体激素后出现进行性小脑综合征。
(2)接受移植后发生的散发性CJD。
3.家族性CJD诊断标准(WHO)
(1)具备上述很可能诊断标准。
(2)一级亲属中有肯定或已很可能患有CJD。
(3)具有神经精神紊乱患者出现特异性PrP基因突变。
4.nvCJD诊断标准(WHO)11①青年发病;②早期表现为焦虑不安、i抑郁、i退隐和其他行为改变等进行性精神异常;③而后数周或数月内出现渐进性小脑症状;④晚期出现记忆力减退甚至痴呆、i锥体外系症状和肌痉挛;⑤EEG无CJD典型特征性变化;⑥个别可出现感觉异常和锥体束症状。
【鉴别诊断】
1.第三脑室肿瘤、i胼胝体神经胶质瘤 虽可有进行性痴呆,l伴有其他局灶性和弥漫性脑病的体征,l但肿瘤的病期较长,l不难鉴别,l加上本病有独特的表现,l如肌阵挛等。
2.Parkinson病 以强直震颤为主而无肌阵挛,l虽然疾病进展到一定时期也存在痴呆的临床类型,l应想到CJD。但Parkinson病的痴呆发生较晚,l且病理学方面无海绵空泡样改变。
3.进行性核上性麻痹 常存在垂直性注视障碍,l如果不存在上视困难则与CJD的肌萎缩型难以鉴别,l前者不仅在临床上表现为广泛的前角细胞受损的体征,l且病理学改变也与运动神经元基本相似,l如加上患者精神状态异常,l两者就更难区别。
4.Alzheimer病典型病例表现为多年的进行性单纯性痴呆,l伴言语困难的局灶性脑病或顶叶损害的体征,l易与CJD区分,l有些Alzheimer病的患者疾病进展较迅速,l又存在显著的运动不能及肌阵挛,l则应与CJD鉴别。但小脑体征的存在有助于排除Alzheimer病,l脑电图检查Alzheimer病不发生周期性放电,lCT或MRI扫描显示Alzheimer病的脑萎缩程度远远超过CJD所见.病理改变还存在Alzheimer病的老年斑和神经纤维缠结,lAlzheimer病尚未见传染。但已有报道CJD与Alzheimer病并存的病例。
(二)库鲁病
库鲁病(Kurn1disease)局限于巴布亚新几内亚EasternHighland省的小范围内,l主要发生在称为Fore的部落人中,l“Kuru”是当地的土族语意为“恐怖性震颤”。Zigas和Gaj-dusek(1957年)首先描述该病,lKlalzo(1959年)报道了该病的病理改变,l并提出与CJD相似,lHadlow(1959年)指出病程与Scrapie相似,l1966年Gajdusek等从Kuru患者脑组织取材注射到黑猩猩脑内,l成功地传播了本病(库鲁病仅能传染猿)。这对揭示人类中枢神经系统疾病的病因和发病机理起重要作用。当地该土族人吃死人肉的习俗可能与疾病传播有关。改变了这一习俗后,l本病就很快得到控制。这项工作具有划时代意义的,l使人类对此类疾病的研究进入一新阶段,lGajdusek由此获得1976年的Noble医学生理学奖。库鲁病的神经症状与羊瘙痒病相似。临床特点主要为行走困难、i步基宽,l相继出现运动障碍、i肌阵挛、i小脑性共济失调、i意向性震颤,l并可有异常不随意动作,l如舞蹈样动作,l手足徐动等,l四肢反射亢进,l锥体束征阳性。晚期进行性痴呆。本病潜伏期4年~30年,l病程4个~24个月,l平均12月。
(三)古斯综合征
首先由Gerstmann于1930年报告,l1981年Masters接种动物发病证实为海绵状脑病病理改变,l近年提取PrP成功,l归类为朊蛋白病。有遗传倾向,l散发者仅少数,l其基因突变位点与家族性CJD不同。目前已知有以下几种突变位点:i密码子102的CCG→CTG(脑氨酸转变为亮氨酸),l密码子117的GCA→GTG(丙氨酸转变为缬氨酸),l密码子198的TTC→TCC(苯丙氨酸转变为丝氨酸),l密码子217的CAG→CGG(谷氨酸转变为精氨酸)。临床特点:i发病年龄26岁~66岁。早期小腿发麻、i疼痛、i感觉异常,l逐渐出现小脑性共济失调,l意向性震颤。脑干受累症状突出,l包括双眼上视不能,l内收不能。双下肢肌无力,l萎缩,l远端感觉减退,l腱反射减退等周围神经病表现。进一步发展可出现精神、i智能障碍。痴呆出现晚而且轻,l可伴有锥体束征和锥体外系征,l晚期可有肌阵挛发作,l以下肢多见。
(四)致死性家族性失眠症
是另一种人类可传递性海绵状脑病,l呈常染色体显性遗传。其突变基因在密码子178,l由天冬氨酸转变为天冬酰氨,l和家族性CJD中某些家系的突变位点相同,l但等位基因突变的异质性可导致不同的临床表现。本病病理改变以丘脑萎缩为主。发病年龄18岁~61岁,l平均49岁,l临床症状为进行性失眠和自主神经功能障碍。睡眠障碍的特征是慢渡和快波睡眠相消失,l伴有幻觉和记忆减退。其他神经系统症状可有共济失调,l肌阵挛、i锥体束征和痴呆。自主神经症状包括多汗、i呼吸和心率增快和发热。脑电图为弥漫性慢渡,l仅1例出现周期性改变。病程1年左右。
二、i检验诊断
朊蛋白病的诊断方法包括组织病理学、i电镜、i生物试验及检测PrPSC等。组织病理学检查是“金标准”,l电镜检查发现异常脑纤维(即瘙痒症相关纤维)的存在可帮助诊断朊蛋白病。生物学试验即将可疑患病动物脑匀浆(或其他组织匀浆)对其他动物(通常用小鼠)进行脑内接种或口服接种,l然后观察被接种动物发病情况,l该方法的敏感性受种属间传播屏障的限制。脑组织活检检测朊蛋白PrPSC是确诊指标。另外,l脑脊液中的一些标志蛋白如脑蛋白14-3-3、iS100蛋白、itau蛋白以及NSE对朊蛋白病也有辅助诊断价值。
【检验项目】
1.脑脊液常规 朊蛋白病患者脑脊液常规检查基本正常。11%的患者细胞数轻微增高,l绝大多数为淋巴细胞。少部分患者脑脊液蛋白轻度增高。
2.朊蛋白PrPSC
(1)检测方法
1)动物传递实验:i是早期判断生物体是否感染朊蛋白、i检测感染滴度、i研究朊蛋白传染性的主要手段。该方法的优点是比较敏感,l是研究朊蛋白生物学特性的重要实验。缺点是费时、i费力,l滴定误差大,l需消耗大量动物,l费用昂贵。由于种属屏障和毒株、i动物个体差异等因素的影响,l传递的成功率往往相差较大,l个别Gerstmann-Straussler综合征(GSS)毒株的动物传递始终未获成功。因此,l实验阴性并不能排除朊蛋白感染.使用转基因动物是一条经济、i实用的削弱或消除种属屏障的实验途径,l可明显改善传递效果。
2)免疫学方法:i免疫学检测方法由于其灵敏度高、i特异性强、i方法众多,l能适合不同检验要求的需要,l目前已成为检测朊蛋白的最主要方法。常用有①组织印迹(histoblot);②斑点印迹(dot-blot);③免疫印迹(immunoblot,lWesternblot);④免疫组化;⑤酶联免疫吸附实验(ELISA)。
3)毛细管SDS-凝胶电泳:i该法标本用量少,l无需免疫学试剂,l判断直观;缺点是需用超速离心机、i专用毛细管电泳仪。
4)荧光标记肽链的毛细管电泳免疫检测法:i是根据免疫竞争原理检测,l采用无区带毛细管电泳技术结合免疫荧光技术,l检测标本中微量的PrPSC。该法灵敏度很高,l能检测到135pg的PrPSC,l可用于检测朊蛋白水平很低的脑外组织(如血液等)。
5)双色强荧光目标扫描法:i是运用共聚焦双色荧光相关分光镜技术检测极其微量的朊蛋白。
6)PrPSC蛋白错误折叠的循环扩增法:i该法是最近发展起来的一种检测微量PrPSC存在的方法,l对组织和体液中用其他方法无法检测到的PrPSC可用该法进行检测,l同时该方法还可确定PrPSC复制是否可在体外产生感染性的独特策略。
(2)标本:i脑脊液、i病变组织、i尿液。
(3)参考范围:i正常人PrPSC阴性。
(4)临床诊断价值和评价:i脑组织活检检测PrPSC是确诊指标。在朊蛋白病的诊断过程中,lPrPSC的存在部位决定了诊断取材。目前已在脑组织、i脊髓、i扁桃体、i脾脏、i淋巴结、i视网膜和近端视神经、i眼结膜、i直肠、i肾上腺、i胸腺、i肾脏及尿液等材料中检测到PrPSC,l其中在淋巴网状组织检测到PrPSC是变异CJD(nvCJD)区别其他朊蛋白病的一个重要标志。由于在nvCJD患者的扁桃体有较高水平的PrPSC存在,l因此扁桃体可作为活检取材部位用以筛查和评价临床前nvCJD感染流行情况。由于在临床症状出现前很长一段时间,l就可在感染患者和动物尿中检测到PrPSC,l因此可通过检测尿中PrPSC而诊断人和动物潜伏期的朊蛋白病。研究还发现,l用尿中的PrPSC接种于仓鼠大脑后。可在270天后都不出现朊蛋白病的表现,l这提示尿中的PrPSC与脑中的不同,l因此,l研究尿中的PrPSC还有助于加深对PrPSC体内代谢的理解。
3.脑脊液中14-3-3蛋白1114-3-3蛋白质是一组真核细胞内高度保守的多功能蛋白质。目前已知至少包括从alpha到theta共7个亚型,l其中zeta亚型mRNA大多克隆自哺乳动物。14-3-3主要分布于脑,l其次是肺、i脾脏和肾脏。在心脏、i睾丸、i胰腺、i卵巢和肌肉含量较少。
(1)检测方法:i蛋白质印迹Westernblot方法。
(2)标本:i脑脊液。
(3)参考范围:i正常人脑脊液14-3-3蛋白质呈阴性。
(4)临床诊断价值和评价:i朊蛋白的沉积是CJD病的主要特征,l脑脊液14-3-3蛋白质的存在,l是CJD的主要分子病理学特征。目前尚不清楚14-3-3蛋白质在CJD中所起的作用及出现在CSF的原因。一般认为是由于大量神经元的破坏,l而导致14-3-3蛋白质漏入到脑脊液中,l14-3-3蛋白质存在的数量可能与神经元破坏比率和数量成正比。14-3-3蛋白质的检测缺乏特异性,l阳性结果还见于疱疹性脑病、i中风的缺氧性腩损伤、i阿尔茨海默病甚至多发性硬化等。14-3-3蛋白质阳性对支持的诊断最有价值,l对于一个临床确诊的病例,l若是阳性则增加了诊断的可信度,l若是阴性,l也不能排除诊断,l14-3-3蛋白质不能被单独用作诊断实验。
4.S100蛋白
(1)检测方法:i酶联免疫吸附试验(ELISA)法。
(2)标本:i血清、i脑脊液。
(3)参考范围:i不同方法有不同的参考范围。
(4)临床诊断价值和评价:iS100蛋白为脑特异性蛋白,l是一个酸性钙结合蛋白,l为两个亚群的同源二聚体或异源二聚体,l分子量分别为10.4kD和10.5kD的α和β蛋白。在脑组织中S100主要存在于胶质细胞中。已有报道脑损伤后血清及脑脊液中S100蛋白浓度增高,l但在这些病例中由于蛋白从肾脏排泄,lS100蛋白浓度又迅速下降,l估计S100蛋白生物半衰期是2h,l最早的研究认为S100蛋白是急性病中脑组织受破坏的标志,l但目前研究表明S100蛋白浓度增高反映星形胶质细胞增生,l而在CJD整个病程中都可见星形胶质细胞增生。因此认为S100蛋白可作为诊断CJD的生化指标。
(5)方法学评价和问题:iELISA法检测S100的浓度,l灵敏度较高、i操作简便、i重复性好,l但试剂盒价格昂贵,l常规应用受到限制。
5.1tau蛋白
(1)检测方法:i酶联免疫吸附试验(ELISA)法。
(2)标本:i脑脊液。
(3)临床诊断价值和评价:itau蛋白是脑内神经元的细胞骨架蛋白之一,l它对稳定微管和神经功能方面起到重要作用。过去认为tau蛋白的异常主要发生于Alzheimer病的老年斑与神经原纤维缠结内、iPick病的Pick小体、i进行性核上性麻痹和皮质基底节变性等神经变性疾病中;而近年从对蛋白质的病理学研究中发现,lCJD患者的脑脊液中tau蛋白水平明显增高,l而且增高程度远远高于Alzheimer病。另外,l脑脊液tau蛋白水平增高,l除见于某些神经变性疾病外,l也可见于急性严重性脑损伤,l如病毒性脑炎、i脑梗死以及脑肿瘤等,l应与这些疾病相鉴别。
6.神经元特异性烯醇化酶(NSE)
(1)检测方法:i时间分辨荧光免疫分析法、i放射免疫法。
(2)标本:i血清、i脑脊液。
(3)临床诊断价值与评价:i患者出现肌阵挛和EEG周期性尖渡时,lCSF中NSE升高最明显,l有人提出CSF中NSE>35ng/ml时,l诊断灵敏度为80%,l特异度为92%。CJD患者早期CSF中神经元特异性烯醇化酶(NSE)的变化相当敏感,l晚期随着神经细胞内NSE的耗竭,l血或CSF中NSE浓度降低。因此,l早期CSF中NSE的检测对判断病情及预后均有实际意义。
7.PrP基因的筛选与分析
(1)检测方法:i聚合酶链反应(PCR)法。
(2)标本:i病变脑组织。
(3)临床诊断价值和评价:i在人类朊蛋白病CJD中有15%为家族性,lGSS突出的特征是小脑共济失调,l与这些疾病相关的则是由宿主编码的一种异常的朊蛋白PrP蛋白在脑组织中沉积,l而编码PrP蛋白的基因位于第二十号染色体,l该基因不同位点的突变可出现不同表现型的家族性朊蛋白病。因此,lPrP基因(PRNP)可作为家族性海绵状脑病的筛选基因。研究表明,l在人的PrP基因129位密码子的多型性(Met或Val)同遗传易感染性有很大关系,l当发生突变(Met→Val)时,l基因为纯合子的个体比杂合子个体易感染朊蛋白病。现已发现正常PrP基因中密码子171位突变(Met→Ser)和219位突变(Glu→Lys)时,l基因型为纯合子的人易患CJD,l而GsS患者中发现密码子102(Pro→Leu)、i117(Ala→Val)、i198(Phe→Ser)和217(Glu→Arg)发生点突变,l使α-螺旋更易转变为β-折叠。PrP从正常主要以α-螺旋的构象转变为以β-折叠为主的构象状态与疾病的发生及其传染性密切相关。
【应用建议】
1.朊蛋白病诊断首选检验项目组合:i组织病理学检查+脑组织PrPSC检测。若组织病理学为脑组织广泛海绵状空泡、i淀粉样蛋白沉淀及神经元退行性改变以及脑组织PrPSC检测为阳性即可确诊。
2.朊蛋白病辅助诊断检验项目建议组合:i脑脊液中14-3-3蛋白+S100蛋白+tau蛋白+NSE。