一、i疾病概述
氨基酸代谢病的代谢障碍主要环节发生在氨基酸本身,l约半数以上能出现精神迟滞或其他的神经系统的功能缺损。按照其发病机制,l本病大体可分为两大类:i
1.酶缺陷类 由于已知的或疑似的某种代谢酶的活性完全或部分缺失所引起的疾病。其中,l按照缺陷的类型而有若干不同的类型:i①酶蛋白本身的缺失;②酶蛋白结构的变化;③以上两型的杂合子;④正常酶蛋白的成熟期延迟;⑤辅因子的缺乏;⑥酶对某些抑制因素的感受性增高。
2.氨基酸转运障碍类11由于氨基酸转运过程中的障碍,l可有一种或多种氨基酸从尿中排出,l其中某些类型还町以影响肠道对氨基酸的吸收过程。另外有一些患者虽然有氨基酸的广泛转运障碍,l但不出现神经系统的症状。
在人体有生理功能的20多种氨基酸中,1已经发现10多种有遗传性的代谢障碍。本章主要介绍几种常见的类型:i
(一)苯丙酮尿症
苯丙酮尿症(phenylketonuria,lPKU)又称苯丙酮酸性精神迟滞或FolEn9病,l是氨基酸代谢病中最常见的类型,l属常染色体隐性遗传,l基因定位于12q24.1,l主要临床特征为智能低下、i痫性发作和毛发稀疏变浅。该病呈全球性分布,l美国人群的发病率为1/111700,l国内11个省市的普查发现其发病率为1/161500。
【病因及发病机制】
本病是起因于肝脏中苯丙氨酸羟化酶(Dhenylalanine1hydroxylase,lPAH)的缺乏,l以致苯丙氨酸(Phe)不能氧化为酪氨酸(Tyr),l影响了各种儿茶酚胺类(多巴胺、i去甲肾上腺素、i肾上腺素等)、i甲状腺素和黑色素的合成。同时使Phe的异常中间代谢产物苯丙酮酸等在体内蓄积,l并且引起了神经系统和全身的各种症状。
【临床表现】
1.患儿出生时多表现正常,l至生后2个月左右出现易激惹、i躁动和呕吐,l有时呈剧烈的喷射性呕吐。至4个~9个月时,l有明显智力发育迟滞.患儿常伴痫性发作。往往发生于18个月前,l可自行消失。在婴儿,l痫性发作可表现为婴儿痉挛,l后期可转变为Lennox型发作或全身型大发作。
2.在体征方面,l患儿肤色较正常儿浅,l毛发稀疏变发黄,l巩膜变蓝,l皮肤粗糙而干燥,l但易于生湿疹,l体表常带有特殊的类似泥土的气味。神经系统的表现除了智能迟滞和抽搐以外,l偶可见有头小、i多动、i震颤、i下肢肌张力轻度减低或增高,l足趾出现中性跖反射或锥体束征等。
3.脑电图常出现高幅失律状态,l单个或多棘波病灶等变化。头颅MRI示T2加权像脑白质中可见高信号,l主要位于颞部及枕部脑室周围,l尤其在大脑中动脉与后动脉血供的交界区。
【诊断】
根据患儿出现严重智能迟滞,l结合皮肤、i毛发等特征,l应考虑本病的可能。可作尿中苯丙酮酸和血浆Phe测定加以明确,l必要时可行脑电图及头颅MRI等协助诊断。应用单型基因及突变基因联合分析,l可有助于检测苯丙酮酸尿症的携带者。
(二)枫糖尿症
枫糖尿症(maple1syrup1urine1disease)是一种家族遗传性脑变性疾病,l由支链氨基酸代谢紊乱所致,l首先由Menkes等发现,l属常染色体隐性遗传。以患者尿中排出的代谢产物具有类似枫糖浆的特异气味为其临床特征,l新生儿的发病率在1/221000~1/401000之间,l我国国内已有报道。
【病因殛发病机制】
正常人体中所具有的三种支链氨基酸是缬、i亮和异亮氨酸,l都不能在体内合成,l而必须从膳食中补充。它们的降解途径是先经过转氨基作用转化为酮基酸,l然后再经过脱羧基作用而逐步分解。现已查明,l本病患儿的体细胞中这三种支链氨基酸的脱羧基酶活性都有明显的减低或缺失.造成体内支链氨基酸和酮酸等在血和脑脊液中蓄积,l同时他们的中间代谢物如α-酮基-β-甲基戊酸(KMVA)和剧异亮氨酸(KMVA转氨基作用后的产物)等也在体内积聚。其中,lKMVA的积聚又可间接地抑制α-羟基酸的分解,l致使α-羟基丁酸和α-羟基异戊酸在病儿的尿中和汗液中大量排泄,l形成特异的枫糖浆气味。
【临床表现】
1.典型病例的临床表现为角弓反张、i肌张力间歇性增高、i痫性发作和大脑功能迅速邀化,l尿液和汗液可有甜枫糖的特殊气味,l部分患者可出现假脑瘤或眼肌麻痹,l约一半患儿有低血糖。
2.在本病的急性期,l头颅MRI可见脑部弥散性水肿,l在大脑深部白质、i内囊后肢、i半卵圆中心后部大脑脚和脑干有典型的重度水肿。
【诊断和鉴别诊断】
根据典型的临床表现,l结合患者尿液或汗液所具有的特殊气味,l便可初步诊断本病,l但确诊必须有血或尿中支链氨基酸的色谱分析,l或是用患儿的白细胞或皮肤成纤维细胞培养,l并直接测定支链氨基酸的脱羧基酶活性。对于基因携带者的检出,l也有使用细胞培养并测定脱羧基酶活性的报告。本病需与新生儿感染、iReye综合征相鉴别。
(三)同型胱氨酸尿症
同型胱氨酸屎症(homocystinurla)又称假性Marfan综合征,l首先由Carson等报道。临床特点有骨骼细长、i眼部异常和常伴发脑血管病等,l尿中出现同型胱氨酸和其他含硫氨基酸。本病为常染色体隐性遗传,l其突变基因可能位于染色体2p,l新生儿的发病率为1/451000。
【病因殛发病机制】
本病系由含硫氨基酸(甲硫氨酸、i胱氨酸)降解代谢障碍,l至少有5种类型的酶缺乏而致。胱硫醚合成酶缺乏,l使甲硫氨酸及其代谢物同型胱氨酸和同型半胱氨酸在体内蓄积,l但维生素B6作为该合成酶的辅酶,l有时可致积贮;N-甲基四氢叶酸同型半胱氨酸甲基转移酶缺乏,l使体内的同型半胱氨酸不能转化为甲硫氨酸,l但大剂量维生素B12有促进此酶活性的作用;N-甲烯四氢叶酸还原酶缺乏,l可致同型半胱氢酸甲基化的能力降低,l使同型半胱氨酸在血中的浓度升高,l甲硫氨酸水平降低;氰钴胺还原酶缺乏可影响多种辅酶的活性。
【临床表现】
1.典型表现①骨骼细长,l尤其是骨骼远端细长,l如蜘蛛状指症,l脊椎后凸或侧凸、i漏斗胸、i弓形足、i椎骨呈“鱼形”等骨骼发育畸形;②智能低下;③眼部异常,l可见晶状体异位;④皮肤可见网状青斑,l面颊部潮红,l毛发稀疏而细;⑤肢带肌肉无力,l以近端为重,l出现肌病步态;⑥内脏器官并发闭塞性血管病,l如肺、i肾和脑的血栓形成和梗死。发生脑梗死者可有偏瘫、i失语和抽搐。
2.肌电图检查 部分患者可出现低幅多相电位肌源性损害。
【诊断】
根据患儿出现智能低下,l眼和骨骼畸形并发肢体肌无力和脑梗死等症状和体征,l结合血清含硫氨基酸水平降低,l尿中同型胱氨酸和同型半胱氨酸水平增高等可予以诊断。对怀疑此酶活性缺乏的胎儿,l可行羊水细胞培养、i酶活性测定作产前诊断。
二、i检验诊断
【一般检验项目】
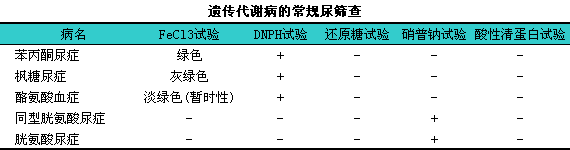
氨基酸遗传代谢病筛查的方法有三氯化铁(FeCl3)试验、i二硝基苯肼(DNPH)试验、i茚三酮试验、i还原糖试验、i硝普钠试验、iCTAB或酸性清蛋白试验,l其临床诊断价值见下表。
【特殊检验项目】
1.尿苯丙酮酸
(1)检测方法:i三氯化铁法。
(2)标本:i尿液。
(3)参考范围:i正常人阴性。
(4)临床诊断价值和评价:i尿液苯丙酮酸阳性见于PKU患者,l但约有25%~50%的病例可能会漏诊。分析中应注意,l该检查方法在枫糖尿症等代谢性疾病也呈阳性反应。故需进一步做血Phe的测定才能确诊。对于新生儿,l如果血Phe的浓度小于900μmol/L,l这两项试验均可呈阴性反应,l故不能用此方法来排除PKU。
(5)方法学评价和问题:i本法灵敏度约100mg/L,l阳性标本可做系列稀释进行粗略定量,l但新生儿在出生后的六周内不易查出,l故于出生六周后检查为宜。检测标本尿液要新鲜,l尿中含有水杨酸制剂、i氯丙嗪及胆红素时,l可呈假阳性。
2.苯丙氨酸
(1)检测方法:iGuthrie细菌抑制法(BIA法)、i干血片荧光定量法(FEIA法)、i高效液相色谱法紫外检测法(HPLC-UV)、i高效液相色普-荧光检测法(HPLC-FLD)。
(2)标本
1)BIA法与FEIA法:i从出生开乳48h~72h以后的新生儿足跟采血。
2)高效液相色谱法:i血清。
(3)参考范围
1)BIA法:i浓度超过标准纸片4mg/dl时,l作重复测定,l如仍超过的,l定为阳性。
2)FEIA法:iPhe≥0.12mmol/L的样本,l应重新采样检测,lPhe浓度≥0.24mmol/L,l并且<1.22mol/L者诊断为高Phe血症;Phe浓度≥1.22mmol/L者诊断为PKU。
3)HPLC-UV法:i正常幼儿血清Phe值(63.2±11.6)μmol/L。
4)HPLC-FLD法:i正常幼儿血清Phe值(92.8±21.3)μmol/L。
(4)临床诊断价值和评价:i血中的Phe岔量对PKU患者的临床诊断有较大的意义,l并且还可以诊断有遗传基因缺陷而无症状的患者,l同时也为产前诊断提供了更为广阔的前景。
(5)方法学评价和问题
1)FEIA法:i该法由于采血量少,l采血方法简单,l样本阴干后可通过邮寄的方式寄到筛查中心进行检测,l较易被患儿家长接受。
2)BIA法:i该法结果直观、i易判断、i操作较简便,l没有很多复杂的环节,l不需特殊的仪器设备,l技术人员比较容易掌握,l适用于大标本量的筛查工作。缺点是试验中引起误差的因素较多,l操作中不仅要控制好温度(过高过低都影响灵敏性),l而且还要掌握平皿之间的厚度(过厚过薄都影响菌环的大小),l就精确而言,l不如化学荧光法好,l只能半定量,l不能为临床提供一个明确的治疗依据。
3)高效液相色谱法:i本法具有灵敏度高、i特异性好、i高重复性、i可定量测定等优点。两种高效液相色谱方法的线性范围宽、i检测限低、i回收率高、i精密度高,l均是PKU筛查、i诊断和治疗监测的良好检测方法,l但HPLC-UV法线性范围宽度、i检测限、i批间CV及标本检测所费时间上更优于HPLC-FLD法。
3.苯丙氨酸和酪氨酸
(1)检测方法:iHPLC-UV、iHPLC-FLD、i质谱分析法。
(2)标本类型:i血清、i血浆。
(3)参考范围:iPhe/Tyr<1.0;Phe/Tyr>2.5可以确诊为PKU。
(4)临床诊断价值和评价:iPKU患者遗传缺陷包括肝脏PAH及其辅因子的合成缺陷,l因此Phe羟化为Tyr的效率降低,l血液中Phe水平升高而Tyr水平下降。如果仅以Phe的浓度为判断指标,l超过90%的新生儿(其Phe浓度>4mg/dl)被证实为假阳性即无PKU。而用Phe与Tyr的比率(Phe/Tyr)则可以在出生后24小时内清楚的区别患儿与非患儿。用此比率可使新生儿出院后不必进行二次筛查,l并且可以减少由过高的假阳性所带来的费用负担。
(5)方法学评价和问题:i现有的用滤纸片采样然后用高效液相色谱柱前或柱后衍生处理再测定的方法简便实用,l免除了标本预处理过程中的诸多变异因素.有效的减少了影响测定结果的诸多环节。
4.四氢生物喋啶
(1)检测方法
1)四氢生物喋呤(tetrahy1drobiopterin,lBH4)负荷试验。
2)高效液相色谱法:i①直接法:i直接测定尿液中的BH4、i新喋呤、i二氢生物喋呤来鉴别经典型PKU与各种类型BH4缺乏症;②间接法:i将尿液处理后,l二氢生物喋啶、iBH4转化为生物喋啶,l再测定生物喋啶、i新喋呤的含量,l求出二者之间的比值及生物喋啶所占的百分比等来鉴别经典型PKU与各种类型BH4缺乏症。
(2)标本:i血液、i尿液。
(3)参考范围
1)BH4负荷试验:i经典型PKU的血Phe对BH4无反应,l血Phe在高浓度水平基本稳定,l24h时平均下降5.3%。
2)尿喋呤谱分析:i经典型PKU1131.69%±24.67%
中度PKU111128.42%±15.92%
BH4缺乏症116.7%±3.23%
(4)临床诊断价值和评价:i由于在PKU筛查阳性的患儿中,l有部分患儿是BH4缺陷所致,l因此及时早期鉴别经典型PKU与BH4缺乏症十分重要。BH4缺乏者在BH4负荷试验中口服BH4片4h~6h,l血Phe浓度下降,l而经典型PKU患者是PAH缺乏,l故对BH4无反应。PKU根据正常饮食下基础血Phe浓度分为:iPhe浓度>11200μmol/L为经典性PKU;Phe浓度在360μmol/L~11200μmol/L为中度PKU;Phe浓度在120μmol/L~36μmol/L为轻度PKU。PKU在服用BH4后24h内血Phe浓度下降至服药前30%或以上,l且排除BH4D后可诊断为BH4反应性PAH缺乏症。
(5)方法学评价和问题
1)试验条件的选择:iHPA患儿血phe浓度≥600μmol/L,l可直接做BH4负荷试验。对已接受低(无)Phe治疗和部分轻中度患儿,l其血Phe浓度<60μmol/L者给予Phe+BH4联合负荷实验,l在试验前先给予Phe(100mg/kg)口服,l服后3h血Phe上升至720~11200μmol/L,l再进行上述BH4负荷试验。用于鉴别BH4代谢过程的还原酶DHPR缺乏所致的BH4缺乏症。
2)BH4负荷试验:iBH4负荷试验不能完全鉴别出DHPR的缺乏型。所以确切的BH4反应性PAH缺乏症和BH4D诊断必须通过尿喋啶谱分析及DHPR测定后确定。服药前和服药后4h~8h留取尿液作尿喋啶谱分析。
3)高效液相色谱法:i本法对于鉴别经典型PKU与BH4缺乏症是十分有效的方法。
5.尿同型胱氨酸定性
(1)检测方法:i硼氧化钠法、i氰化硝普钠试验(Brand反应)、i硝普银法、i硝普银显色法。
(2)标本:i尿液。
(3)参考范围:i正常人阴性。
(4)临床诊断价值和评价;胱氨酸尿症是一种先天性代谢疾病,l尿中出现胱氨酸结晶,l可引起尿路复发性胱氨酸结石。正常人为阴性。阳性见于胱氨酸尿症患者。
(5)方法学评价和问题
1)氰化硝普钠法与硼氢化钠法:i尿液Brand反应是初筛同型胱氨酸尿症的定性试验,l该方法虽然简便快速,l但尿中如有酮体可造成假阳性,l胱氨酸尿症和其他含硫化合物对结果也有干扰作用.故该试验不是诊断同型胱氨酸尿症的特异试验。
2)硝普银法:i对同型胱氨酸具有特异性和灵敏性,l其最小检出值为2.0mg/100ml。将该方法与薄层层析结合起来,l方法更为灵敏与特异,l其最小检出值为1.0mg/100ml。主要优点是可根据标准氨基酸的Rf值推断待测氨基酸,l可同时进行定性与半定量分析,l是诊断同型胱氨酸尿症的一种值得推荐的方法。
6.蛋氨酸负荷试验
(1)检测方法:i口服L-蛋氨酸1.6g或DL-蛋氨酸1.6g,l于服药后0h、i2h、i4h、i8h、i16h,l各取静脉血,l分离血清。同时分别收集各时期尿液进行层析。根据标准蛋氨酸和同型胱氨酸的Rf值找出各样品中蛋氨酸和同型胱氨酸的斑点,l并按照各色斑的深浅和大小与标准色斑比较以半定量各样品中蛋氨酸和同型胱氨酸的含量。
(2)标本:i血清、i尿。
(3)参考范围:i正常人仅在负荷L-蛋氨酸后2h有微量增加,l但曲线很快下降。而DL-蛋氨酸负荷试验正常人的蛋氨酸在负荷后2h明显升高,l4h后才迅速下降并趋于正常水平。
(4)临床诊断价值和评价:i同型胱氨酸尿症患者在服用1.6g1L-蛋氨酸或1.691DL-蛋氨酸后血中蛋氨酸和同型胱氨酸均明显增加,l并在较长时间内不能回复。尿中蛋氢酸和同型胱氨酸的排泄量在服药后均明显增高。在4h左右达最高峰,l以后缓慢下降,l16h仍不能回复到服药前水平。但正常人的蛋氨酸在负荷后2h明显升高,l4h后才迅速下降并趋于正常水平。此可能因为负荷DL-蛋氨酸后正常人尿中排泄的蛋氨酸是机体处理D-蛋氨酸不完全,l过多的部分从尿中排出之故,l但与患者相比仍然较少,l下降趋势亦快。L-蛋氨酸负荷试验与DL-蛋氨酸负荷试验两者大致相同,l同型胱氨酸尿症患者与正常人的不同是患者不仅尿中排泄较多的蛋氨酸,l而且存在大量同型胱氨酸,l这对诊断本病十分重要。
【应用建议】
1.对本病初筛检验项目:i三氯化铁(FeCl3)试验、i二硝基苯肼(DNPH)试验、i茚三酮试验、i还原糖试验、i硝普钠试验、iCTAB或酸性清蛋白试验。
2.确诊实验:i血、i尿苯丙酮酸。
3.其他项目可以辅助诊断。